
In this era there is growing interest in rare diseases and more investigations are focused on their molecular background. By advanced molecular techniques and global sharing of specialistic knowledge and clinical data, an immense collection of genetic irregularities has been elucidated in association with human disease. Molecular knowledge can be helpful in further classifying and diagnosing diseases, especially rare phenomena.
Concerning genetics, DNA molecules with their substantiating nucleotides are the building stones of chromosomes and deliver the basis for cellular function. A better understanding of rare diseases like CMTC (Cutis Marmorata Telangiectatica Congenita) by comparison of DNA sequences seems to be within arm’s reach. However, still, a complex puzzle has to be solved before patients concerned can take the advantages of those investments.
Purpose of this article is to make clear what recent developments in genetic research mean in the light of disturbances in cellular function. The scientific article of Jordan M (ref. 1) is taken as a starting point because herein corresponding DNA mutations are highlighted within the chromosomes of patients, most of whom show aberrant skin phenomena combined with under- or overgrowth features as is seen in CMTC too. A summary of this article can be found elsewhere on the CMTC-website (ref. 2). In short, the key protein which appeared to be mutated in all of the cases analysed is the Gα protein, which is part of a trimeric GPCR (G protein-coupled receptors). The subunits β and γ remain out of scope here.
Above the genetics, diverse signalling pathways are considered here which could be disturbed by such DNA mutations. Signalling via a molecular cascade (like a domino) is the way by which processes within the cell are communicated and controlled. Primary and secondary messengers play an important role as intermediate molecules. The environment of cells is changing continuously, therefore receptors are located at the outer side of the cellular membrane with the function of a kind of sensor, in order to trigger signal transduction when necessary. A pathway can end up in the nucleus of a cell, where it influences the production machinery of newly build proteins or interact with other pathways or molecules to be activated.
This insight within the cell mainly illustrates the complexity of molecular mechanisms. This can be a small step in the direction of understanding a rare disease with phenomena as shown in patients with CMTC.
CMTC is not a clearly delineated syndrome and can overlap with other, related syndromes, like Sturge-Weber syndrome or Adams-Oliver syndrome (ref. 3). Another disorder which shows overlapping symptoms with CMTC is Cutis Marmorata (CM), being a broader occurring phenomenon which is not diagnostic (ref. 3).
Furthermore, the diversity of this skin-related syndrome becomes clear when a Google search is performed in order to find out what the phenomena you may encounter look like. In short, for the description of CMTC phrases are used like ‘marbled-looking skin’, or ‘fixed patches of mottled skin’, or ‘discolored patches of skin caused by widened surface blood vessels’, or ‘reticular vascular skin pattern’, or ‘distribution of skin lesions’ and so on. Besides, musculoskeletal anomalies resulting in body asymmetry together with other phenomena are characteristic for this rare syndrome (ref. 4).
Since a few years a subcategory of macrocephaly-capillary malformation (M-CM) can be distinguished from CMTC as another syndrome but likewise potentially resulting from a genetic mosaicism (ref. 3). In a mosaicism only part of the cells constituting a tissue – like skin – is affected while the other part is not. In short a simplified explanation of CMTC in the context of genetic mosaicism could be as follows (outlined box).
As CMTC is a sporadically occurring, thus rare congenital disorder it is thought that during early embryonic development a postzygotic (so-called ‘de novo’) mutation occurs within the genome that influences subsequent cellular function. From that point on, during the following growing phase new cells are generated from this particular cell by cellular division. Thus chromosomes are copied and taken over, including the DNA mutation. Thus, the mosaic pattern can arise that is obvious in the newborn’s skin (showing actually the underlying vascular defects (ref. 2). Also other defects can have arisen from this DNA mutation and in turn more than a single mutation may be involved.
In short, the manifestations of CMTC, belonging to the diverse group of vascular anomalies, are complex, with varying tissues being affected. This could explain the diversity of descriptions of the syndrome and reclassification in the past few years, having consequences for the nomenclature too. Moreover, abbreviations used in some cases can be more confusing than clarifying.
Switching from the phenotypical perspective to the yet unexplored area of genotypes could help in strengthening the diagnosis of patients and acquiring insight in the basis of cellular mechanisms being involved in this kind of syndrome.
In the last decennium research activities using ‘next generation sequencing’ (NGS) have led to DNA mutations which are typically found in the affected tissue(s) of patients with rare, skin-related syndromes. No mutations are associated with CMTC in particular yet.
A recent study described by Jordan et al. in 2019 focused on cases with ‘undiagnosed cutaneous anomalies’. Twelve out of the 32 cases demonstrating capillary malformations showed a Cutis Marmorata- (CM-) type of skin anomaly.
The researchers studied two genes, the Gα11 and the GαQ gene, both coding for another subtype of G protein α. They have found that the cases with a CM-type were significantly associated with a mutation within its occupant Gα11 gene.
Likewise, the ipsilateral hypotrophy phenomenon was associated with a Gα11 mutation. Moreover, in all but one cases the mutation concerned the amino acid arginine (Arg) at position 183 within the protein sequence, either in Gα11 (Arg183Cys or Arg183His or Gln209His) or in GαQ (Arg183Gln; Table 1).
Performance of this study was referred to as ‘reversed genotyping’, pointing to an attempt of assigning genotypic characters to phenotypic phenomena within the cases investigated. In each of the cases, unaffected tissue was used as an internal control.
Summarizing:
(1) Jordan distinguished two groups of cases, according to their respective Gα11- or GαQ mutation.
(2) The CM-type of skin anomaly was associated with Gα11. On the contrary, Sturge-Weber type of disorders was not associated with either Gα11- or GαQ mutations. G11 and Gq are related proteins both belonging to the Gq class of G proteins (ref. 4).
(3) From these results arginine can be assumed to play an important role in their function as primary messenger molecule concerned with GPCR binding.
(4) The internal control tissue lacking the specific DNA mutation within their corresponding cells is in accordance with the concept of genetic mosaicism
| Amino acid affected by mutation | Number of cases | G protein |
|---|---|---|
| Arg183Cys | 7 | GNA11 |
| Arg183His | 1 | GNA11 |
| Gln209His | 1 | GNA11 |
| Arg183Gln | 3 | GNAq |
As Cutis Marmorata is considered as a symptom instead of a syndrome (ref. 3), extended research has to make clear if this kind of DNA mutations in Gα11 will show up in CMTC lesions too.
The marbled skin predominantly characterizing CMTC, with telangiectasia only applying to a minority of patients, strengthens this hypothesis. Greene et al. already suggested alterations affecting the MEK pathway (including GPCR, the G protein-coupled receptor) in capillary malformations (ref. 5). Only the gene ARL6IP6 is known in relation to specific CMTC cases (ref. 4).
An association with GαQ mutation has been found earlier in the Sturge-Weber syndrome. In the Adams-Oliver syndrome on the other hand mutations in ARHGAP31 (a monomeric type of G-protein) are recognized as also in 5 additional genes that can account for this syndrome Ref. 4,6).
In contrast to mutations in the PIK3CA spectrum (as in the related Klippel-Trenauney syndrome), mutations in the GαQ/11 spectrum of G proteins are known to account for more diffuse syndromes (ref. 5). Nevertheless, also capillary malformations as in CMTC are affiliated under an umbrella, bearing the “G-protein receptor” mutations and named as “GNA-vascular anomaly” (GNAVAS) (ref. 5). With the closely related GαQ/11/14 genes currently being the main contributors.
A cell is provided with several kinds of receptors, among which the G-protein coupled receptor (GPCR), by which it can communicate with and adapt to environmental changes. GPCRs are located across the cellular membrane and can be triggered by a ligand binding at the outside. In response, a signal is sent to the inner side of the receptor, where it can subsequently bind and activate the primary effector molecule, here the G protein α* (Figure 1; αGTP in green). At the end of the pathway, when the signal is transduced, calcium (Ca2+) is released and protein kinases** C and D (PKC,D) are activated, subsequently phosphorylating and thus activating RAF and other proteins.
By this, several physiological processes within the cell are regulated, especially of relevance during embryonic development.
* The cascade is initiated by the action of the G protein α which in turn activates phospholipase C-β (PLC-β, a lipid cleaving enzyme) to activate the second messengers DAG and IP3, influencing several functions within the cell.
** Many activation steps constituting a cascade are effected by the phosphorylation of proteins by protein kinases.
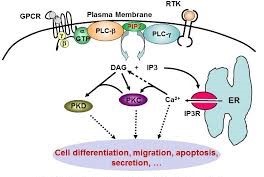
Figure 1 – Signaling pathways of IP3 and DAG generation (ref. 7)
Besides the cleavage of PIP2, Gα can also activate the PI3K pathway, at the end of the phosphorylation cascade resulting in MAPK activation. This is an example of the crosstalk between different cellular pathways initiated after ligand binding to GPCR (Figure 2).
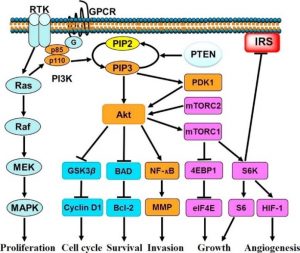
Figure 2 – PI3K stimulation of cell growth. PI3K, phosphoinositol-3-kinase; GPCR, G protein-coupled receptor; RTK, tyrosine kinase receptor (ref. 8).
Editor Dr. Marjon ten Hoor-Suykerbuyk