
In de afgelopen 25-30 jaar is de kennis over de oorzaak van vasculaire malformaties enorm toegenomen. Er zijn 11 genen gevonden waarin mutaties optreden die verantwoordelijk zijn voor het ontstaan van een malformatie. Ook zijn er remmende stoffen gevonden die een gunstig effect hebben op het verloop van veel van deze aandoeningen. Miikka Vikkula* geeft in zijn lezing een beeld van wat er aan kennis is bijgekomen door genetisch en klinisch onderzoek en hoever we nu zijn.
Vasculaire malformaties is eigenlijk een spectrum van ziekten, dat betekent dat er veel verschillende soorten zijn (>100). Onder de verschillende groepen malformaties, bijvoorbeeld veneuze malformaties (VM), zijn er weer veel verschillende subgroepen.
Vele vasculaire malformaties zijn zeldzaam, maar niet allemaal. Bijvoorbeeld cerebrale caverneuze malformatie (CCM) is niet zo zeldzaam (1/200) maar slechts weinig mensen die het hebben, hebben klinische symptomen. Behandelingen zijn vooral gericht op het verlichten van de symptomen, zoals laser, chirurgie, sclerotherapie, embolisatie, compressie. De oorzaak van de ziekte wordt met deze behandeling echter niet weggenomen.
Vasculaire malformaties werden vroeger meestal angioma’s genoemd. Aangezien er zoveel verschillende soorten angioma’s waren was het dus lastig om te begrijpen waarover men het had. In 1982 maakten Mulliken en Young daarom een indeling (classificatie) van de verschillende ziekten op basis van het stromingstype (fast/slow flow) en het vaattype (ader/slagader/lymfevaten). Deze classificatie heeft zich verder geëvolueerd naarmate er meer kennis kwam over de verschillende aandoeningen.
Vasculaire malformaties kunnen erfelijk zijn maar ook niet-erfelijk. De erfelijke vormen zijn erg zeldzaam, maar het onderzoek in families met een erfelijke vorm heeft enorm geholpen om de mutaties in genen op te sporen die een rol spelen in het ontstaan van deze aandoeningen. In 1996 werd het eerste gen (TIE2) ontdekt dat verantwoordelijk is voor een vasculaire malformatie (https://pubmed.ncbi.nlm.nih.gov/8980225 ). Vikkula deed toen onderzoek in Boston (Harvard Medical School). Daar kwam hij op het spoor van dit gen dat verantwoordelijk is voor een veneuze malformatie in een grote Italiaanse familie. Inmiddels zijn er elf genen gevonden voor verschillende aandoeningen. Het laatste gen (CM-AVM2/EPHB4-gen) werd in 2017 gevonden.
Vikkula vond het eigenaardig dat de laesies alleen op bepaalde plekken op het lichaam voorkwamen. “Als de mutatie in alle cellen aanwezig is bij de familiaire vorm, waarom is het effect ervan dan alleen op bepaalde plekken aanwezig? Om het zo gelokaliseerd te krijgen heb je twee mutaties in hetzelfde gen nodig, dachten we. Een in de maternale kopie en een in het vaderlijke gen exemplaar. Er is dus een tweede treffer (somatische mutatie) nodig.”
Die hypothese bleek te kloppen uit onderzoek aan Glomuvenous malformations (GVMs) dat in 2002 gepubliceerd werd. (Brouillard et al, Am J Hum Gen: https://pubmed.ncbi.nlm.nih.gov/11845407/). Een van de individuen uit een familie met GVMs bleek naast de erfelijke mutatie ook een mutatie te hebben die later in zijn leven moet zijn opgetreden (somatische mutatie). Ook bij andere erfelijke vormen van vasculaire malformaties is dit fenomeen gevonden. Maar niet bij alle malformaties. Het kan zijn dat de tweede mutatie (nog) niet is gevonden.
Opmerkelijk was ook dat niet alle mutaties in de genen tot functieverlies leidden: één (in het TIE2-gen) leidde tot een gain-of-function (te veel functie). Later werd ontdekt dat in 60% van de niet-erfelijke veneuze malformaties er een somatische mutatie is in TIE2 die functiewinst oplevert. In de loop van de jaren werden nog meer mutaties ontdekt: een AKT-mutatie in het Proteus syndroom in 2011, een PIK3CA-mutatie in het CLOVES-syndroom in 2013. Een GNAQ-mutatie in capillaire malformatie en Sturge Weber-syndroom enz. Dit heeft onze kijk op vasculaire anomalieën volledig veranderd.
De meeste vasculaire malformaties zijn niet erfelijk. Maar soms bootsen de erfelijke letsels de niet-erfelijke na (nb. PHTS). Dus genetische counseling is wel noodzakelijk, want als het een erfelijke vorm is, kunnen andere familieleden het ook hebben en risico lopen. Hoe beter de genetische analyse, hoe beter de diagnose van de ziekte en de follow-up/behandeling.
De verschillende ziekten kunnen nu worden aangeduid met de namen van de genen waarin mutaties zijn opgetreden. Bij vasculaire anomalieën zijn dat bijvoorbeeld vaak TIE2 en PIK3CA. Een mutatie in het PIK3CA-gen kan verantwoordelijk zijn voor zeer verschillende fenotypen van de ziekte. Het kan bijvoorbeeld macrodactylie en syndactylie 2-3 en CM van de rechtervoet van CLOVES veroorzaken maar ook CLVM van de linker thorax, KT-syndroom en gemengde LM van de linker thorax veroorzaken. Verschillende uitkomsten kunnen worden veroorzaakt door hetzelfde gen. Daarom is het belangrijk om een correcte beschrijving (fenotype) van de ziekte te hebben samen met een genetische diagnose.
10 van de 11 genen hebben een mutatie die een verlies van functie veroorzaakt. Maar dit functieverlies zit meestal in een inhibitor, zodat een pathway (zie figuur Theranostics) niet wordt geremd en er een toename van de functie is van het eiwit aan het eind van de pathway. Men zou dus kunnen zeggen dat de meeste mutaties tot nu toe tot een toename van de functie in de pathways veroorzaken.
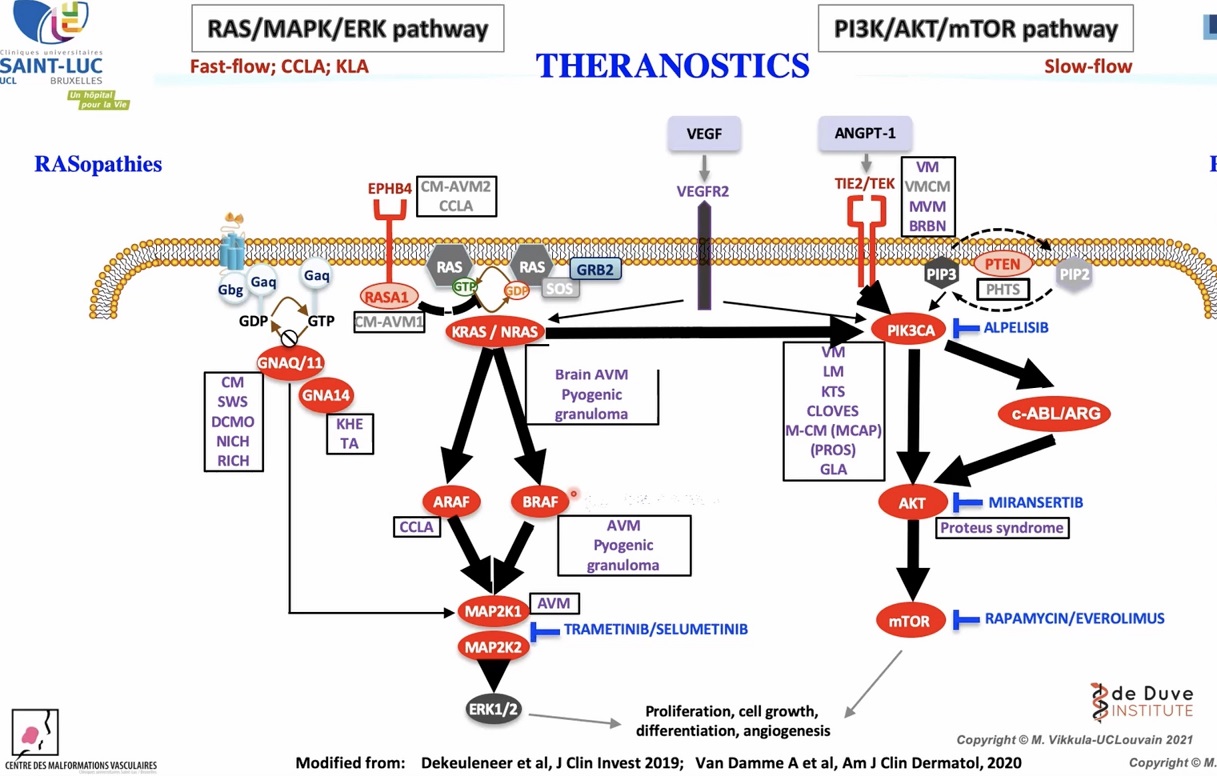 Weergave van alle belangrijke routes die een rol spelen in de regulering van metabolisme in zoogdieren. Rood (namen van genen), paars (namen van ziekten) en blauw (namen van inhibitoren).
Weergave van alle belangrijke routes die een rol spelen in de regulering van metabolisme in zoogdieren. Rood (namen van genen), paars (namen van ziekten) en blauw (namen van inhibitoren).
Mutante cellen (weefsel van geopereerde patiënten) kunnen worden gekweekt, zodat hun ontwikkeling in het laboratorium te bestuderen is. Vergeleken met normale humane endotheelcellen gekweekt in het laboratorium groeien gemuteerde cellen veel meer boven op elkaar. De normale cellen hebben een keienachtig (cobblestone) uiterlijk. Het toevoegen van rapamycine en alpelisib aan de mutante cellen had een gunstig effect.
Om de ziekte in een levend model te bestuderen heeft men een diermodel gecreëerd. Daarvoor injecteerden onderzoekers mutante cellen in een immunodeficiënte muis (J Clin Invest 2015, Boscolo E. et al (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4588237/). Een immunodeficiënte muis heeft geen afweercellen en reageert dus niet op de ingespoten cellen. Na verloop van tijd integreren de cellen in het weefsel van de muis. Het weefsel van de ‘gemuteerde’ muis lijkt erg op het weefsel van patiënten met een VM. Er ontstaan blauwe laesieplekken. Vier dagen na toediening van rapamycine stopte de groei van de laesies volledig.
Rapamycine wordt nu bij mensen getest. Het is al gebruikt bij patiënten voor andere ziekten (kanker), dus het is relatief veilig om bij patiënten te gebruiken. In een fase IIA klinische studie werden 3 patiënten met een TIE2-mutatie en 3 patiënten met een PIK3CA-mutatie met verschillende klinische fenotypes behandeld met rapamycine. Zij waren reeds behandeld met chirurgie en sclerotherapie. Alle patiënten hadden baat bij de behandeling met rapamycine, de biologische parameters verbeterden, de functionele stoornissen waren minder ernstig. Dit was het bewijs van het concept: we kunnen moleculaire therapie (inhibitoren) gebruiken bij vasculaire malformaties.
Rapamycine wordt gebruikt in verschillende klinische studies en het wordt ook off-label gebruikt door talrijke artsen over de hele wereld. Er loopt tevens een kleinschalig klinisch onderzoek naar miransertib en twee onderzoeken naar alpelisib. Daarnaast worden inhibitoren voor de MAP2K-route, trametinib en selumetinib getest. Deze remmers zijn ook gebruikt bij kanker omdat deze genen ook gemuteerd zijn bij kanker.
Preklinische modellen zijn dus belangrijk om de details van de ontwikkeling van laesies te bestuderen en om geneesmiddelen te screenen. Gedetailleerde klinische proeven zijn nodig om veilige precisiegeneesmiddelen voor vasculaire anomalieën te ontwikkelen.
Vikkula benadrukt ook weer eens het cruciale belang van internationale samenwerking op het gebied van zeldzame ziekten. Het VASCERN-VASCA-netwerk is zeer belangrijk. Ernstige en moeilijke gevallen worden besproken. Er worden richtlijnen opgesteld voor patiënten en er worden webinars gegeven. Er is ook een VASCERN-app. Handig wanneer je op reis bent en er is een noodgeval. De app helpt je snel een goed medisch centrum in de buurt te vinden.
* Prof. dr. Miikka Vikkula: Human Genetics, Institut de Duve, Brussel
Dit verslag van Vikkula’s lezing is geschreven door dr. Lilian Vermeer